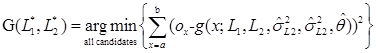Genotyping of microsatellite loci
GenoTan
GenoTan

|
Genotyping of microsatellite loci
GenoTan
GenoTan
|
|
IntroductionGenoTan is a free tool to identify length variation of microsatellites from short sequence reads. Inferring lengths of inherited microsatellite alleles with single base pair resolution from short sequence reads is challenging due to several sources of noise including PCR amplification errors, individual cell mutation, misalignment or mis-mapping caused by the repetitive nature of the microsatellites. We have developed a method using a discretized Gaussian mixture model combined with a rules-based approach to identify inherited variation of microsatellite loci from short sequence reads, which effectively distinguishes length variants from INDEL errors at homopolymers. License: GPL V3 (http://www.gnu.org/licenses/gpl-3.0.html)
How to use GenoTan Table of Contents 1. Discretized Gaussian mixture distribution 2. Searching repeats of short motifs Realignment of sequence reads mapped to the microsatellite loci
Methods used in GenoTan1. Discretized Gaussian mixture distributionGenoTan uses a discretized Gaussian distribution to address homopolymer errors from short sequence reads mapped to microsatellite loci. Since probabilities of insertion and deletion errors in a sequence are proportional, they can be calculated from the cumulative distribution function of the Gaussian distribution. Let lL be
the length of a candidate allele L at a target locus and let x be
the observed length of the microsatellite sequence with INDEL errors in a read
mapped to the locus with an assumption in which the length x is derived
from the original length lL. Let FL(t)
and f L(t) denote the distribution and the density
functions of a Gaussian random variable with mean lL and
variance
where x=0,1,2,... ,
and
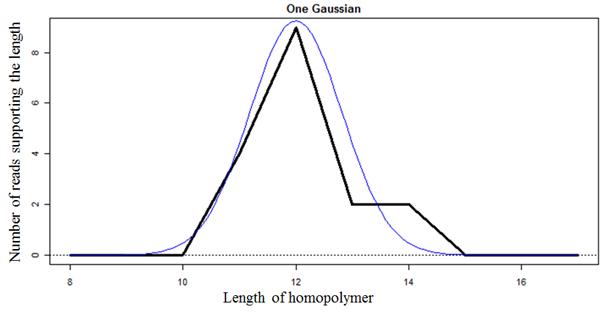
Recurrence of INDEL errors in sequence reads. The reference is a genomic sequence of Brucella suis 1330 which is a prokaryote with a haploid genome, and the locus is a unique region in the genome. The sequence reads were generated from the Illumina 101 cycle paired-end sequencing technology. Insertions or deletion errors by sequencing errors or other sources, such as PCR amplification error or individual cell mutation, results in various lengths of G homopolymers in sequence reads.
Gaussian distribution to express the bidirectional INDEL errors in sequence reads. The graph shows a Gaussian distribution for the locus at the previous figure. The chances of insertion errors and deletion errors are proportional.
For the heterozygous loci with allele lengths, lL1 and lL2, we can use the mixture distribution of (1) as follows
, where
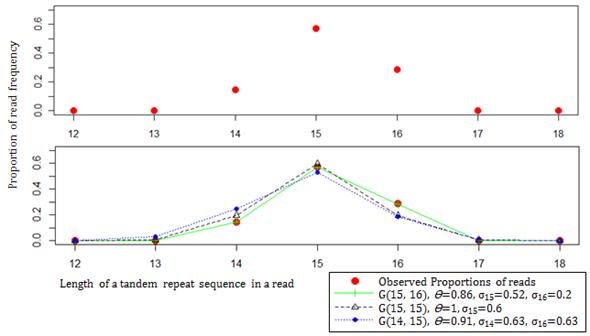
Heterozygous fitting. Graphs represent the process of heterozygous fitting for 4 and 6 mapped reads containing microsatellite sequences with lengths 13 and 16 bases respectively at a locus.
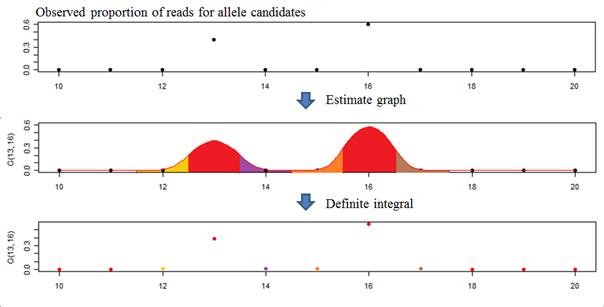
If the sequence reads mapped to a same microsatellite locus contain INDEL errors, the number of observed lengths of the microsatellite at the locus would be equal to or more than 2. Since the inherited alleles are unknown, all observed lengths can be allele candidates. Then we can apply the g(x) function for each combination of two allele candidates (two of the same candidate for a homozygous genotype), calculate the squared error of each combination, and select the allele pair, L1*and L2*, that generates the minimum squared error as follows
, where ox is an observed proportion of reads containing a length x microsatellite sequence, a is the minimum observed length minus a fixed amount k, and b is the maximum observed length plus k, where k is set to be five as default value. This is necessary because the g(x) function generates output values for all possible sequence lengths, the comparison between observed proportions and expected proportions need to be extended beyond the minimum and maximum observed lengths. So, the boundaries of the calculation are extended by an additional value k. As an example, suppose
that we have 2, 8 and 4 mapped reads containing microsatellite sequences with
lengths 14, 15 and 16 bases respectively at a locus. The list of possible
genotype candidates G(lL1, lL2) for
the locus are G(14, 14), G(14, 15), G(14, 16), G(15,
15), G(15, 16), and G(16, 16). In the example, the
observed minimum and maximum lengths are 14 and 16 respectively, and the
observed and expected values from the equation 3 are compared for x
ranging from 9 to 21. While the observed ratio of read counts between the
highest read frequency allele (lL1=15) and the
second highest read frequency allele (lL2=16) is
0.5 (=4/8), the read ratio of those two alleles estimated by the NLS function
was 0.163 (= (1-
2. Decision of genotypingThe most probable genotype for a given sequence reads mapped to a locus is decided by equation 3, but it is bias toward calling heterozygous genotypes, because the Gaussian mixture model is better fit to the training data when more distributions are mixed. However since reads supporting one or both predicted alleles may be from noise including individual cell mutation, PCR amplification error, sequencing error and mismapping, an evaluation method is necessary. We use a rule-based
approach to choose alleles and to decide the homozygosity of each locus because
the frequencies of INDEL error reads derived from mis-mapping, PCR amplification
error and individual cell mutation are more difficult to measure than that from
the sequencing error. For this approach, we assign a confidence score to each
allele instead of calculating the probability of a genotype (a two allele set) for
a locus. The probability of each allele can be generated by the equation 1 as pL1(lL1)
or pL2(lL2) if we assume the
read frequencies from two different alleles at the heterozygotic locus are not
correlated. However DNA fragments from two paired chromosomes have the same
chances of being sequenced and the read frequencies of two alleles would be
tend to be similar. If the proportion of reads for an allele candidate Llow
with lower read frequency is too small compared to that for another allele
candidate Lhigh with higher read frequency, we may conclude
that the reads for the allele candidate Llow are from noise
and the locus is homozygous. Considering this condition, we multiplied ratio of
In the final results, an allele candidate from the predicted genotype is removed when its confidence score is lower than given cutoff values (we set 0.35 for Lhigh and 0.25 for Llow). When a confidence score of Llow is lower than the cutoff value, GenoTan generates a partial genotyping call for the locus in which only one allele is called while the other allele is reported as unknown. It only reports the genotype of the locus as homozygous when the number of reads supporting the selected allele is more than 4 and its confidence score is equal to or higher than 0.9. The confidence score of the second allele, Lhigh2, at a homozygous locus is calculated by
where [0.5n] represents the probability of that another unobserved allele exists when n reads support the selected allele.
Getting startedGenoTan is available at the Download page and is distributed with three additional PERL programs including makeListFromTRF.pl, searchTandemRepeats.pl and realignReadsOnRepeats.pl.
InstallationThe current release requires GSL (http://www.gnu.org/software/gsl, developed with GSL 1.14) and runs in the LINUX environment.
$ tar xzf genotan.[VERSION].tar.gz $ cd genotan.[VERSION]
If the GSL library was installed in a personal directory, edit the 'Makefile' file in the directory. Ex) If the GSL library was installed in the '$(HOME)/opt' directory,
INCLUDES= -I. -I$(HOME)/opt/include LIBPATH= -L$(HOME)/opt/lib
Change '$(HOME)/opt/include' to a full path of a directory containing GSL headers and '$(HOME)/opt/lib' to a full path of a directory containing GSL libs. Then, type 'make' to compile it.
$ make
Searching microsatellitesWe provide two methods to search microsatellite sequences. The first method is to use TRF (Tandem Repeat Finder, http://tandem.bu.edu/trf/trf.html) which searches imperfect repeats of short motifs from reference sequences. The second method is to search perfect repeats of short motifs from reference sequences directly.
1. Using TRFTo convert the output of TRF to the input file of GenoTan, we provide makeListFromTRF.pl.
$ trf reference.fa 2 7 5 80 10 14 6 -h $ [GenoTan_path]/makeListFromTRF.pl -r reference.fa -t reference.fa.2.7.5.80.10.14.6.dat -o mic.lst
2. Searching repeats of short motifsWe also provide searchTandemRepeats.pl to search repeats of short motifs from reference sequences directly.
$ [GenoTan_path]/searchTandemRepeats.pl reference.fa -o mic.lst
Realignment of sequence reads mapped to the microsatellite lociMisalignment of sequence reads mapped to the repeat sequences is frequently observed. Since GenoTan does not have a process to realign sequence reads, its results significantly rely on the alignment quality. To reduce the misalignment problem we provide realignReadsOnRepeats.pl which is a realignment program for the sequence reads.
$ [GenoTan_path]/realignReadsOnRepeats.pl -r reference.fa -m mic.lst -s input.sam -o realigned.sam
The sequence reads can be also realigned by GATK (Genome Analysis Toolkit, http://www.broadinstitute.org/gsa/wiki).
$ cat mic.lst | perl -ne '@arr=split/\t/; print "$arr[0]:$arr[1]-".($arr[1]+$arr[2]-1)."\n";' > mic.intervals $ java -jar GenomeAnalysisTK.jar -T IndelRealigner
-R reference.fa -targetIntervals mic.intervals -I input.bam
Run GenoTan
$ [GenoTan_path]/genotan -m mic.lst realigned.sam -o genotype.txt
Usage :
genotan -m <microsatellite list file> <sam or bam file> [-o <output
file>] [-L off]
1. Input filesGenoTan takes a microsatellite list file (which requires '-m') and a SAM or BAM file containing mapped reads. The microsatellite list file has the following format.
[chromosome], [starting position], [length], [motif], [sequence] in a tab-delimited format
Ex) chr1 26454 12 GT GTGTGTGTGTGT chr1 28589 15 T TTTTTTTTTTTTTTT chr1 44836 32 AAAT AAATAAATAAATAAATAAATAAATAAATAAAT chr1 722365 20 ATTT ATTTATTTATTTATTTATTT
2. Other options-L off : Turn off the normalization of read counts using the allele lengths (default : on) -C FLOAT [0.01-0.99] : If a confidence score of a higher read frequency allele at a locus is lower than the cutoff value, a genotype of the locus will not be reported. (default : 0.35) -c FLOAT [0.01-0.99] : If a confidence score of a lower read frequency allele at a locus is lower than the cutoff value, the allele will not be reported. (default : 0.25) -q INT [0-255] : if a mapping quality of a read is lower than the cutoff value, the read will not be used. (default : 5) -O INT [0: no sequence, 1: sequence output]: It decides the output style. (default : 1)
3. Output fileEx)
The output file of GenoTan is a tab-delimited file. Each line consists of
'ext. del. in flanking left:right' : Lengths of deletions adjoined to the microsatellite locus. For example :
reference : ATTCGAGGGATATATATATATCAAGTAC read 1 : ATTCGAG----ATATATATATCAAG read 2 : CGAG----ATATATATATCAAGTAC
In this example, the length of the microsatellite in the reference and the length of the allele candidate in the reads are 12 bases and 10 bases respectively. But there are two additional deleted bases in the left flanking sequence of the microsatellite. To separate the deletions in the repeat sequence from that in the flanking sequences, the allele is displayed as '10(2:0)' in the output file.
4. Test with simulated dataThe simulated data is available online [Download]. It was created from human genome data with artificial microsatellite sequences of 1~8-mer motifs. Repeat sequences in sequence reads for the microsatellite loci were generated by the Binomial random function (p=0.5) and two Gaussian random functions. The inherited alleles (artificial) for each locus are in the 'test.allele.lst' file. There are three different BAM files and each file contains reads mapped to the microsatellite loci in different coverage.
$ [GenoTan_path]/genotan -m test.mic.lst test.cov.10.bam -L off -o genotype.cov.10.txt
|